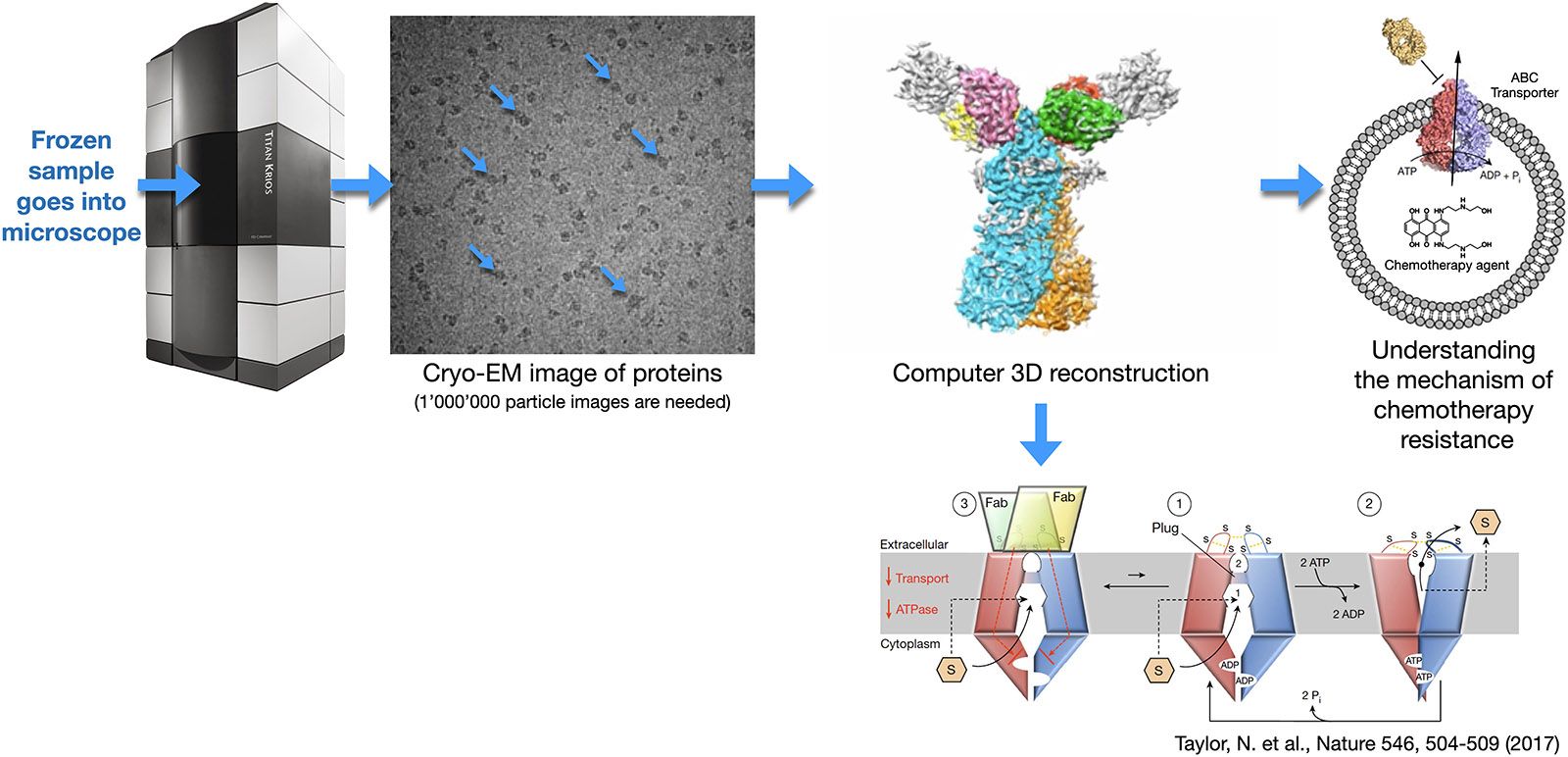
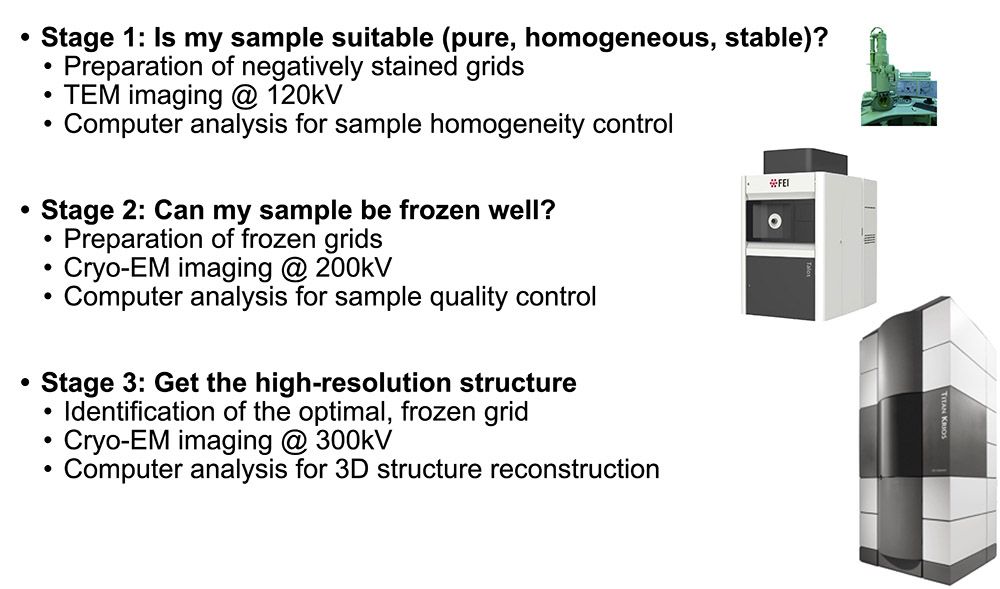
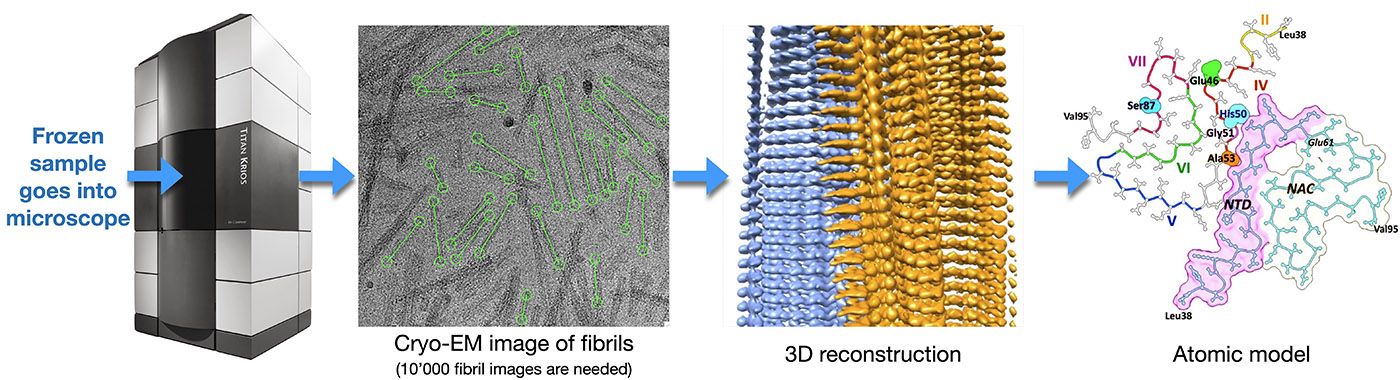
This method is used to determine the high-resolution three-dimensional (3D) structure of isolated protein complexes. Samples are flash frozen in a thin layer of vitreous ice and imaged at cryogenic temperatures by transmission electron microscopy (TEM), e.g., using the TFS Titan Krios microscope. The direct electron detectors (DEDs) of these instruments greatly increase the achievable image contrast, and allow ‘movie mode’ imaging. In this mode, a series of dose-fractionated images (i.e., movies that are containing 40 to 100 frames) of suitable sample regions are recorded. The movie mode data collection allows sample drift to be detected and corrected. After the required alignment, the individual frames are merged to increase image contrast, i.e., to improve the overall signal-to-noise ratio.
The Falcon4 detectors employed by the DCI alternatively allow recording image data as a series of X/Y electron impact coordinates, together with the time point of the arrival of the electron. Such "electron event recordings" can later be interpreted by software and transformed into movies of variable pixel resolution and a variable number of frames for a movie.
Each sample region imaged contains many protein complexes oriented in different ways. Their images, i.e., projections in 2 dimensional space (termed 2D projections) are matched, classified and reconstructed in a procedure called ‘single particle analysis’ to obtain the 3D structure of the complex. Alpha helices and beta sheets can be visualized by this method, and, since the introduction of DEDs, atomic resolution is frequently achieved.